Blog
See all posts14/12/2021 - Journal Club
How to choose a proper combination of search engines to maximize Peptide and Protein Identification?
While going through our personal projects during our journey in PROTrEIN (Training of computational proteomics researchers), we were able to see the importance of the database search engines and post-processing in the analyzing of proteomics mass spectra data. The choice of select combined search engines and post-processing methods are critical. Hence the choice of our paper titled ‘Optimization of Search Engines and Post-processing Approaches to Maximize Peptide and Protein Identification for High-Resolution Mass Data’.
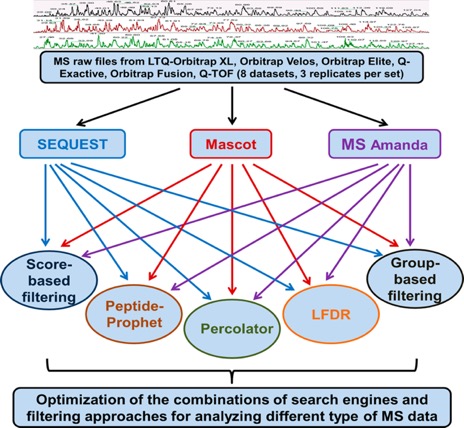
Chengjian Tu at al. published the paper in 2020, where they did 360 analysis investigation the performance of three popular search engines (SEQUEST, Mascot, and MS Amanda) in conjunction with five filtering approaches, including respective score-based filtering, a group-based approach, local false discovery rate (LFDR), Peptide Prophet, and Percolator. A total of eight data sets from various proteomes (e.g., E. coli, yeast, and human) produced by various instruments with high-accuracy survey scan (MS1) and high- or low-accuracy fragment ion scan (MS2) (LTQ-Orbitrap, Orbitrap-Velos, Orbitrap-Elite, Q-Exactive, Orbitrap-Fusion, and Q-TOF) were analyzed. It was found combinations involving Percolator achieved outperformed more peptide and protein identifications at the same FDR level than the other 12 combinations for all data sets. The combination of percolator with SEQUEST, and with MS Amenda outperformed on low accuracy MS2 (ion trap or IT) and high accuracy MS2 (Orbitrap or TOF). Also did experiment without using peculator, SEQUEST-group performs the best for MS2 dataset produced by collision-induced dissociation (CID) and IT analysis, Mascot-LFDR give more identifications for dataset generated by higher energy collisional dissociation and analyzed in Orbitrap (HCD−OT) and in Orbitrap Fusion (HCD−IT), MS Amanda−Group excels for the Q-TOF data set and the Orbitrap Velos HCD−OT data set. This combinations without peculator guidelines for combinations of selecting methods.
The workflow details are shown in Figure 1, as eight data sets (three replicates per data set) generated from three organisms (E. coli, yeast, and human) by various MS instruments including LTQ Orbitrap XL, Q-TOF, Orbitrap Velos, Orbitrap Elite, Q-Exactive, and Orbitrap Fusion were investigated, with the three popular search engines representing different characteristics (SEQUEST, Mascot, and MS Amanda (i.e., SEQUEST, emphasis on similarity, Mascot, emphasis on probability, and MS Amanda, emphasis on high-accuracy MS2 spectra)) were selected. Meanwhile, the five most popular filtering approaches (original-score-based filtering, group-based, PeptideProphet, Percolator, and LFDR) were assessed for each search engine.
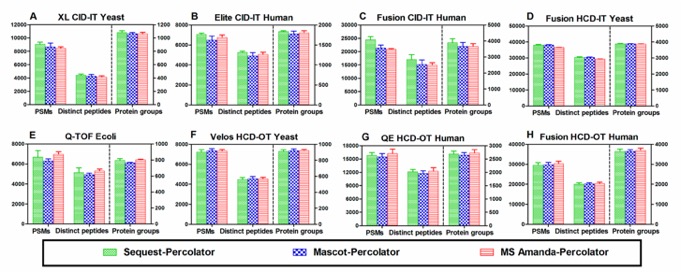
Due to not suitability of naïve score-based filtering for every dataset the LFDR method introduced for estimations of PSMs using SEQUEST, which use confidence level Investigation by Bayesian approach, which improves identification for higher mass accuracy data. The SEQUEST with percolator significantly outperformed than other four methods in PSMs, distinct peptides, and protein groups of all the data sets analyzed. In the case of distinct peptides, the 21.6% of total identified peptides are unique to SEQUEST-percolator, by comparison, only less than 1% of peptides are unique to each of the other four combinations.
The Mascot-score resulted in the lowest number of identifications at the same protein FDR level, Percolator outperformed than other four filtering methods in the numbers identified PSMs, distinct peptides, and protein groups in all the data sets at a protein FDR of 1%. The data sets use for experiments, Mascot−Percolator provided, respectively, 34.6 to 175.9%, 31.9 to 154.0%, and 8.6 to 68.3% more PSM, distinct peptides, and protein groups compared to the Mascot−score approach. In the case of distinct peptides, when using the Mascot database search, Percolator and LFDR provide the best results for all data sets with low- or high-accuracy MS2 spectra.
In the terms of the number of PSMs, distinct peptides, and proteins identified from low-accuracy by MS Amanda-score perform lower than SEQUEST-score or Mascot score, these leads to the MS Amanda-score not suitable for low-accuracy detector. From the analysis of high-resolution and high-accuracy MS2 data for MS Amanda, the Percolator outperformed than the other four postprocessing methods. MS Amanda− Percolator provided an average of 27.9%, 27.6%, and 13.1% more PSM, distinct peptides, and protein groups.Instead of using original score-based filtering approaches, the percolator afforded the best performance out of the popular postprocessing approaches, used in these studies. As previous studies, SEQUEST and Mascot are algorithm design for HCD, ETD, and CID fragmentation with high- or low-accuracy MS2 spectra while MS Amanda is optimal for high-accuracy MS2 data. As shown in Figure 2, The results of combination of percolator with different search in terms of PSMs, distinct peptides, and protein groups, while SEQUEST−Percolator slightly outperformed Mascot−Percolator and MS Amanda−Percolator for the CID−IT data sets. Interestingly, although MS Amanda is designed for high-accuracy MS2 data, when Percolator was used, its performances for both high- and low-accuracy data sets are similar to those of the other two search engines, which is contrary to the findings when the score-based method is used. Even though MS Amanda is not optimal for CID−IT data, MS Amanda−Percolator achieved the better performance than Mascot or SEQUEST coupling with the other four filtering approaches (score-based, group-based, PeptideProphet, and LFDR), which once again indicated that Percolator is a better processing approach.
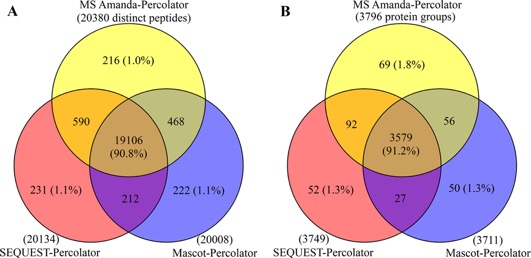
As shown in the Venn diagrams in Figure 3, superb overlap among the three search engines coupled to Percolator was observed, 90.8% at the peptide level and 91.2% at the protein level. Moreover, only ∼1% of distinct peptides were unique to each of these three combinations, and more than 96% of peptides or proteins were commonly identified between two of the combinations. After comparison with previous studies combination of multiple search engines, conclude as the percolator approach minimizes the preference and bias by different searching algorithms and thereby permits more confident identification.
Reference: doi: 10.1021/acs.jproteome.5b00536 J. Proteome Res. 2015, 14, 4662−4673

Categories
Latest posts

08/05/2024 - Journal Club
Cross-Border Collaboration: Enhancing Peptide Identification with MS2Rescore and MS Amanda

08/09/2023 - Journal Club
Exploring Cellular Complexity: Unveiling Single-Cell Proteomics

23/08/2023 - Journal Club
Modeling Lower-Order Statistics to Enable Decoy-Free FDR Estimation in Proteomics