Blog
See all posts17/02/2022 - Journal Club
How to train your modified peptide MS/MS spectrum predictor?
This month’s PROTrEIN journal club covers an article presenting a tool – pDeep2 – capable of predicting MS/MS spectra of modified peptides.1 pDeep2 is built using a machine learning technique that makes it possible to generate prediction even when there are only a few datasets available for training the model. But… What is this machine learning technique? What are modified peptides? And…- What do they have to do with the PROTrEIN international training network? Read along and you’ll get those answers just in a few minutes!
Background
A short intro to PTMs
The number of protein-coding genes is estimated to be ~20 000 but the collection of human proteoforms is remarkably more diverse than that. There are numerous contributors to the variations at DNA, RNA and protein levels. On the DNA level the main source of variations are coding single-nucleotide polymorphisms (cSNPs) and mutations, on the RNA level it’s alternative splicing and on the protein level post-translational modifications (PTMs).2,3
There have been over 400 PTMs discovered already and their number is growing. PTMs can affect protein function, for instance, phosphorylation4 is the reason for many signalling cascades, acetylation shows relation to blood pressure and hormone regulation, neurodegenerative diseases 5,6, glycosylation is involved in many cellular processes, such as the formation of biofilms and the antimicrobial resistance of various pathogens7, ubiquitination of a proteins most commonly results in the degradation of the proteins 8 and the list goes on.
Because of the aforementioned impacts, it’s clear that identifying and gaining more knowledge about PTMs carries a huge relevance in cell biology and it has great biomedical importance. The main challenges regarding their analysis lie in their usual low abundance, their complexity and the fact that not only their presence but their exact location on the backbone peptides is important too.9,10
Spectra prediction & identifying modified peptides
Why to predict MS/MS spectra?
The most common way of peptide identification is done with database search. Database search consists in comparing experimental spectra to theoretical spectra but traditionally it works with uniform intensities. Another method for peptide identification is spectral library search that uses previously acquired and annotated spectra. Spectral library search includes intensity information but libraries are limited to “already seen” peptides. MS/MS spectra prediction can provide the intensity dimension to theoretical spectra using only some basic inputs such as the peptide sequence and collision energy. Using these predictions, database identification scores can be refined, the number of identification can be increased and false identification can be decreased.
MS/MS spectra prediction is nowadays built on deep learning methods, borrowing a lot of ideas from Natural Language Processing. The most common neural network architectures applied in spectra prediction are built on Long short-term memory (LSTM) and recurrent neural networks. While spectra prediction has its own difficulties, by including PTMs in the process, further challenges emerge. Predictor neural networks are trained using experimental datasets and even though there are numerous annotated datasets for peptides without modifications, the number of such datasets for modified peptides is limited.
Machine learning strategies for the small dataset problem
Publicly available datasets can be quite scarce when we try to find solutions to our proteomics related problems. Well annotated datasets are not always available therefore different strategies need to be applied to create useful machine learning models. Some of these strategies are choosing simpler models, careful feature selection, combining several models, extending datasets by either creating synthetic samples or pooling data from other possible sources and finally applying transfer learning.
Transfer learning attempts to create methods to transfer knowledge learned in one or more source tasks and use that knowledge to enhance learning in a similar task. Simply put, it means training a universal model on available large data-sets and then reusing and adapting the model to another task with a small available dataset. These pre-trained models are likely to give better predictions than models only trained with the small available datasets and they are working especially well with deep learning methods.11,12
Short summary of the paper
This month’s article of the PROTrEIN Journal club presents a machine learning model developed using transfer learning to predict MS/MS spectra for modified peptides. The methods and results demonstrated in the paper can be a use for the members of the ITN and for anyone who is reading this blog post.
Introduction
The goal of the authors was to address the problem of training a good machine learning model that is capable of predicting spectra not only for unmodified peptides and peptides with common PTMs but for low-abundant PTMs as well. With their previous model called pDeep they showed it is possible make accurate spectra predictions for unmodified peptides and they used pDeep2 – a faster and more flexible version of pDeep – as basis to develop a model able to predict spectra for modified peptides.
Methods
Regarding features for the model, the researchers used a one-hot indicator vector with dimension 20 for each amino acid and a chemical composition vector with dimension 8 to represent common PTMs. These features are then concatenated with precursor charge state, instrument type and collision energy. The outputs of the model are the relative intensities of b/y ions with +1 and +2 charge states. Their initial unmodified pDeep2 model had 2 hidden bidirectional dynamic LSTM (Bi-Dy-LSTM) layers with a hidden layer size of the LSTM cell 256. The dropout was set to 0.2 with 100 epochs, mini-batch size of 1024 and a learning rate of 0.001. The selected loss function for the training was mean absolute error.
After training the first accurate pDeep2 model using data sets of unmodified PSMs, the authors moved on to the transfer learning step. The first Bi-Dy-LSTM layer was fine-tuned to fit the new input PTM features and the output layer was tuned to fit new outputs but the intermediate hidden layers stayed frozen. The researchers developed the PTM version of pDeep2 so it can also consider b/y ions with neutral losses (NLs) of PTMs. The learning parameters for the transfer learning were set to 20 epochs with a mini-batch size of 1024 and learning rate of 0.001 using mean absolute error as loss function.
The authors used Pearson correlation coefficient (PCC) as a similarity metric to compare the predicted spectrum with the experimental one. P(PCC > x) or PPCC>x was used as a criterion to evaluate the performance of predictions. PPCC>x refers to the proportion of PCCs that is greater than a given value x. For example, PPCC>0.75 = 95% means there are 95% PCCs greater than 0.75.
Results
The unmodified pDeep2 model was trained and tested on a total of ∼8 000 000 high-quality unmodified peptide-spectrum with the result that PPCC>0.75 was higher than 90% and PPCC>0.90 was higher than 80%. The researchers tested multiple transfer learning methods and found the “tune-first-last” method best suited for their purposes. The data sets they used for fine tuning included sets of ordinary MS runs that included common PTMs such as oxidation on Met and data sets of 21 synthetic PTMs. The pre-trained model was then fine tuned with aforementioned different PTM data sets and it’s prediction performance was compared to the pre-trained, non-transfer trained and combined models – where combined means the model was trained by combining the unmodified and modified data. The transfer trained models outperformed all other models and reached remarkable performance (See figure below).
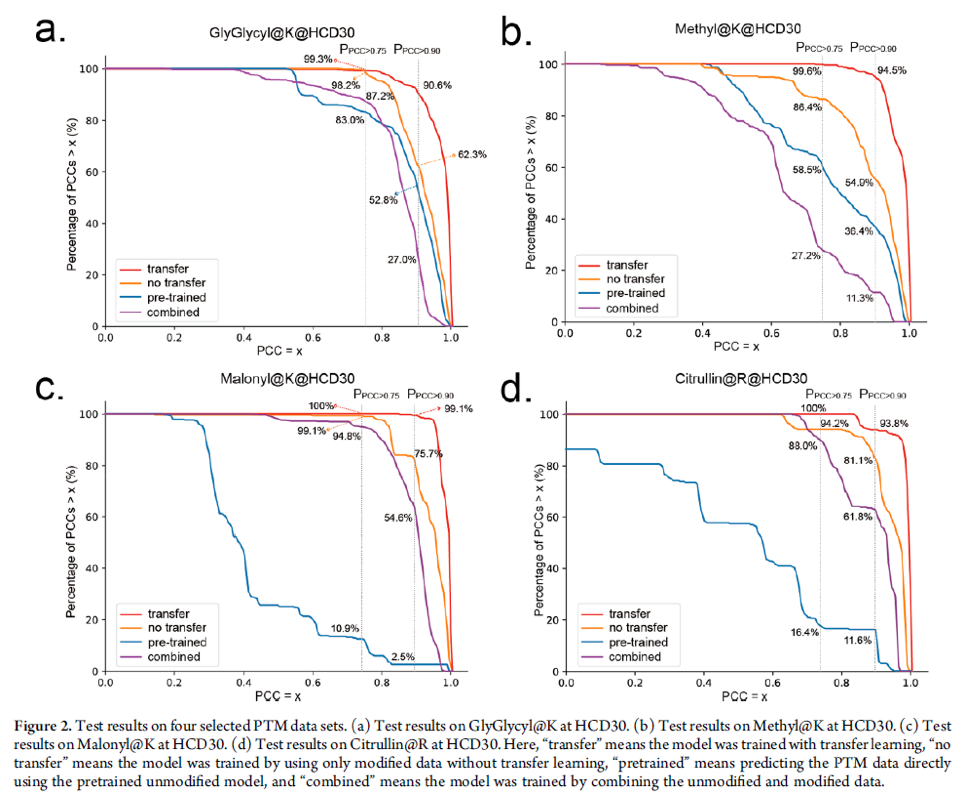
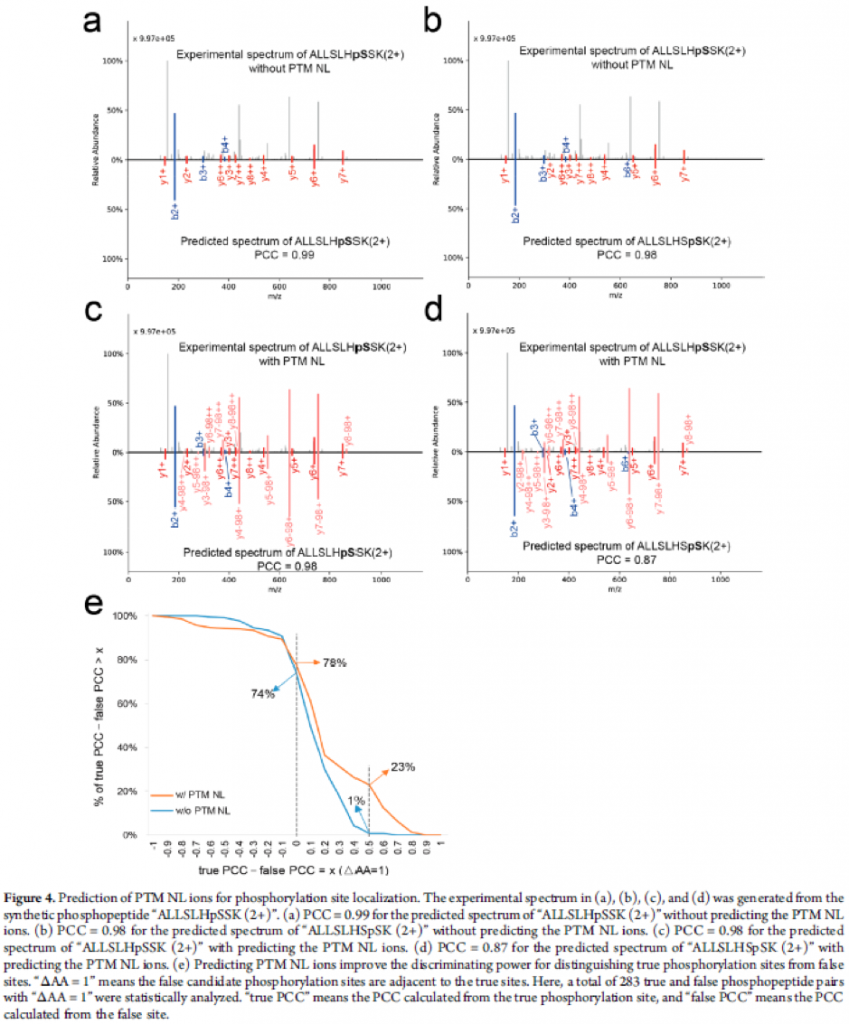
The authors also presented that the prediction of PTM NL fragment ions of phosphopeptides may provide complementary information for localising the PTM sites. While PCCs of true and false phosphorylation sites for a sequence were almost the same without PTM NL fragment ions, they managed to show difference when PTM NL fragment ions were included in the model. (See figure below)
Conclusions
As discussed before, the relevance of PTMs in bio-medicine is huge, therefore they have received a lot of attention from researchers in the proteomics field. In this blog, we briefly covered the topics of PTMs, spectra prediction and transfer learning. We presented the article “MS/MS Spectrum Prediction for Modified Peptides Using pDeep2 Trained by Transfer Learning“1 from Zeng et al. shortly with the intent to spark interest towards these topics and give some useful insights. This study is another example showing that, thanks to the great advancements in machine learning, it is now possible to use deep learning to solve proteomics problems.
The PROTrEIN international training network’s goal is to train bioinformatics researchers in the field of mass spectrometry based proteomics, therefore it is not a surprise that the PhD topics reflect that interest towards PTMs and the use of machine learning. More than one third of the PROTrEIN ITN projects involve modified peptides and/or machine learning. These projects linked to PTMs cover a whole range of different topics such as PTM landscape characterisation, predicting modified peptide behaviour, identifying and quantifying modified peptides. Other programs are focused on using machine learning more broadly to improve and add new features to tools like MaxQuant and PROSIT.
We hope you enjoyed this post and will check back to us next month!
Bibliography
1. Zeng, W.-F. et al. MS/MS Spectrum Prediction for Modified Peptides Using pDeep2 Trained by Transfer Learning. Anal. Chem. 91, 9724–9731 (2019).
2. Aebersold, R. et al. How many human proteoforms are there? Nat. Chem. Biol. 14, 206–214 (2018).
3. Ramazi, S. & Zahiri, J. Post-translational modifications in proteins: resources, tools and prediction methods. Database J. Biol. Databases Curation 2021, baab012 (2021).
4. Ardito, F., Giuliani, M., Perrone, D., Troiano, G. & Muzio, L. L. The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review). Int. J. Mol. Med. 40, 271–280 (2017).
5. Xia, C., Tao, Y., Li, M., Che, T. & Qu, J. Protein acetylation and deacetylation: An important regulatory modification in gene transcription (Review). Exp. Ther. Med. 20, 2923–2940 (2020).
6. Drazic, A., Myklebust, L. M., Ree, R. & Arnesen, T. The world of protein acetylation. Biochim. Biophys. Acta BBA – Proteins Proteomics 1864, 1372–1401 (2016).
7. Schulze, S. et al. Enhancing Open Modification Searches via a Combined Approach Facilitated by Ursgal. J. Proteome Res. 20, 1986–1996 (2021).
8. Guo, H. J. & Tadi, P. Biochemistry, Ubiquitination. in StatPearls (StatPearls Publishing, 2022).
9. Torres, M. P., Dewhurst, H. & Sundararaman, N. Proteome-wide Structural Analysis of PTM Hotspots Reveals Regulatory Elements Predicted to Impact Biological Function and Disease *. Mol. Cell. Proteomics 15, 3513–3528 (2016).
10. Su, M.-G. et al. Investigation and identification of functional post-translational modification sites associated with drug binding and protein-protein interactions. BMC Syst. Biol. 11, 132 (2017).
11. Torrey, L., Shavlik, J., Walker, T. & Maclin, R. Transfer Learning via Advice Taking. in Advances in Machine Learning I (eds. Koronacki, J., Raś, Z. W., Wierzchoń, S. T. & Kacprzyk, J.) vol. 262 147–170 (Springer Berlin Heidelberg, 2010).
12. 7 Effective Ways to Deal With a Small Dataset | HackerNoon. https://hackernoon.com/7-effective-ways-to-deal-with-a-small-dataset-2gyl407s.

Categories
Latest posts

08/05/2024 - Journal Club
Cross-Border Collaboration: Enhancing Peptide Identification with MS2Rescore and MS Amanda

08/09/2023 - Journal Club
Exploring Cellular Complexity: Unveiling Single-Cell Proteomics

23/08/2023 - Journal Club
Modeling Lower-Order Statistics to Enable Decoy-Free FDR Estimation in Proteomics