Blog
See all posts15/04/2022 - Journal Club
Adding the third dimension to protein modifications analysis
Short description
In the study of post-translational modifications from mass spectrometry experiments, proteins are almost always represented as a linear string of amino acids. However, it should be kept in mind that proteins’ function is conferred by their 3-dimensional structure and that understanding the modifications of proteins calls for studying them in their structural context. In this month’s PROTrEIN journal club, we present a method that aims to bridge the gap between protein structure and data from mass-spectrometry experiments in the context of proteins modification studies.The paper we chose is “The structural context of PTMs at a proteome-wide scale” (Bludau et al. 2022). We selected this paper as it demonstrates how the interpretation of data in mass-spectrometry can be taken a step further by leveraging the knowledge provided by the recent advances in structural proteomics.
Introduction
The regulation and fine-tuning of cellular processes are possible because proteins are highly dynamic molecules. Once synthesized proteins undergo many changes that impact their properties and the way they interact with other proteins or molecules. The alteration of proteins’ properties can be induced in many ways such as changes in the environment with variations in pH or temperature for instance. Protein activity can also be modulated by other proteins with the addition or removal of chemical groups to protein. These modifications of proteins that occur after their synthesis are referred to as post-translational modifications (PTMs). Most proteins are highly modified and to this date, more than 150 different types of PTMs have been discovered. The PTMs are at the core of the regulation of many cellular processes. As an example, the modification of a protein by the addition of ubiquitin is a marker for protein degradation as it is recognized by the proteasome (Hochstrasser et, 1996). A not-to-be-missed topic when it comes to PTMs is the modification of histones. Histones are proteins that bind with the DNA to form a complex referred to as chromatin. Chromatin is a dynamic structure that heavily influences the accessibility of transcription factors to the DNA and therefore genes. The histones are known to be heavily modified and deciphering what is the effect of the different histones’ modification on chromatin structure is crucial to the understanding of epigenetic mechanisms (Millán-Zambrano et al, 2022).
Measuring protein modifications with mass spectrometry
Several techniques are used to study post-translational modification. For example, immunoprecipitation enables to target and measure proteins with a specific modification or set of modifications. Mass-spectrometry techniques allow for the large-scale study of these post-translational modifications. In a mass spectrometry experiment, it is possible to measure the entire protein sequence for many proteins simultaneously. This allows for a global study of changes in PTMs pattern but also raises many challenges in the interpretation of the data.
Linking proteins modification and structure
The objective of a PTMs study is often to identify a specific protein’s PTM or PTM pattern that can be linked to the regulation of a cellular process. Because of the numerous modifications that are present on proteins, it is particularly difficult to determine which modifications are possibly involved in a given cellular process.As proteins are folded into a 3-dimensional structure not all parts of the protein are exposed to the same extent. The function of a specific modification most likely depends on the localization of that modification in the protein conformation. Protein structural information can therefore be included in PTMs studies to better understand and pinpoint PTMs that have a strong regulatory role. Some regions of the protein fold into a stable structure when some other regions do adopt any stable conformation, these unstable amino-acid chains are referred to as intrinsically disordered regions (IDRs). IDRs are regions of proteins that are frequently involved in protein-protein interaction, that are often enriched in post-translational modifications (see animation below).
In the paper presented here, the authors set out to explore the structural context in which PTMs take place, with the goal of understanding the functional role of the PTMs based on their position on the 3d structure.
The protein folding problem
Despite the advances in technology, the experimental definition of a single 3d structure might take up to several years. For this reason, a different paradigm was explored with the aim of determining the 3d structures starting from the sole amino acids sequence of a protein, the so-called ‘protein folding problem’. Over the course of the past years, various computational approaches have been developed for this goal, using methods ranging from molecular simulations to evolutionary approaches. While the approaches based on molecular interactions rely on computationally heavy procedures that become intractable with the growing size of proteins, recent techniques based on machine learning have been proven to be much more effective for predicting the structure of longer peptide sequences.
CASP and AlphaFold2
The CASP experiment (Critical Assessment of protein Structure Prediction) is recognized as the state-of-the-art method for evaluating the predictive capability of algorithms aimed at solving the protein folding problem. Every two years, the most advanced methods are tested on a set of completely new and previously uncovered protein structures that have been experimentally determined.
During the past CASP14 assessment, held in 2020, the AlphaFold2 approach (Jumper et al. 2021), developed in Google DeepMind’s laboratories, proved to be remarkably effective for solving the protein folding problem. AlphaFold2 is based on an end-to-end deep learning pipeline that incorporates biological knowledge with attention-based methods for graph inference. This architecture obtained considerably low root-mean-squared deviation when predicting the atomic position of the backbone’s carbon atoms. AlphaFold2 enabled the creation of a complete database (https://alphafold.ebi.ac.uk/) containing almost 1 million accurate protein structure predictions (Varaldi et al., 2021).
Although AlphaFold2 constitutes a huge step toward the advancement of structural informatics, it does not provide a way to introduce PTMs information inside the models. We chose therefore to present a paper that leverages the knowledge that AlphaFold2 provided, with the aim of integrating PTM information with the protein structures provided.
“The structural context of PTMs at a proteome-wide scale” (Bludau et al. 2022)
Determining IDRs and side-chain exposure from AlphaFold2 dataset
While the experimental information on protein folding is often limited to specific regions of the sequences, relying on the AlphaFold database enables access to extended information on the folding of complete proteins. However, to exploit the provided 3d structures for PTM analyses, some preprocessing steps are needed in order to generate local information on the shape of the proteins. For each amino acid, the most interesting features to be extracted are its side chain exposure and its belonging to either a structured region or an intrinsically disordered one (IDR).
In order to determine the side chain exposure of a specific amino acid, the authors define a metric called prediction-aware part-sphere exposure (pPSE). The pPSE counts the number of alpha carbon atoms that are present in proximity of each amino acid, by considering a half-sphere centered in the amino acid itself and directed towards its side chain. The radius of the spheres is based on the average size of amino acids and on the flexibility of the side chains. However, in order to take into account the uncertainty introduced with the predicted 3d structures, the radius value is adjusted for each amino acid based on the prediction alignment error (PAE) provided by AlphaFold2. Each residue in the sequence can therefore be associated with a value of pPSE: the lower the PSE, the more the other amino acids are distant from the one under consideration, and the more its side chain will be exposed to other interactions.
The authors also proved that pPSE can be used to predict IDR regions in an accurate way. For the specific goal of annotating IDRs, the whole sphere is considered when computing the pPSE values, in order to disregard the orientation of the side chain, and the pPSE values are smoothed by computing a sliding average along the amino acid sequence. This metric proved to be the most effective in retrieving IDRs when comparing the results to the available information on already annotated proteins.
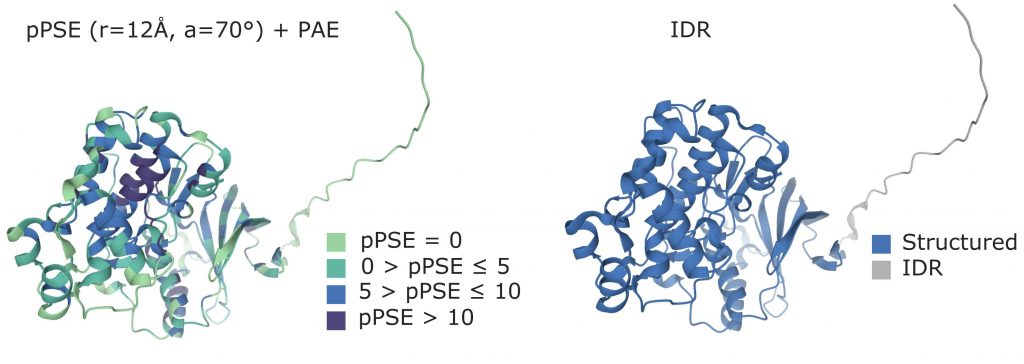
Figure from (Bludau et al., 2022).
Are PTMs localized in IDRs or structured regions?
Through enrichment analyses, the authors observed that most PTMs, including phosphorylations, seem to appear in IDRs. The same holds true for acetylation when only the sites with known regulatory regions are considered, while they seem to be underrepresented when considering all PTM sites. Ubiquitination appears instead to be enriched in structured regions, especially when these regions are not associated with regulatory functions. The hypothesis presented by the authors, and verified in different experimental conditions, is that ubiquitination in structured regions could be the main driver that causes misfolding of proteins. The misfolding subsequently leads to exposure of normally unexposed regions where the proteasome can bind and proceed with the degradation of the protein. When the PTMs are instead located in IDRs, the authors observe that these modifications are usually enriched in short IDRs that are included between two larger structured sequences. An enrichment analysis of the proteins with modifications in short IDRs highlighted processes such as ATP binding, transmembrane activity and protein kinase activity. The authors observe a significant overlap between short IDRs and activation loops of kinases, which are known to be subject to structural changes when phosphorylated. Particularly, the majority of the kinases that show overlap between the annotated activation loop and the predicted short IDR, also show regulatory functional phosposites in the same region. These findings show the importance of studying PTM sites in correspondence of short IDRs, as they seem to be directly correlated with functional regulation.
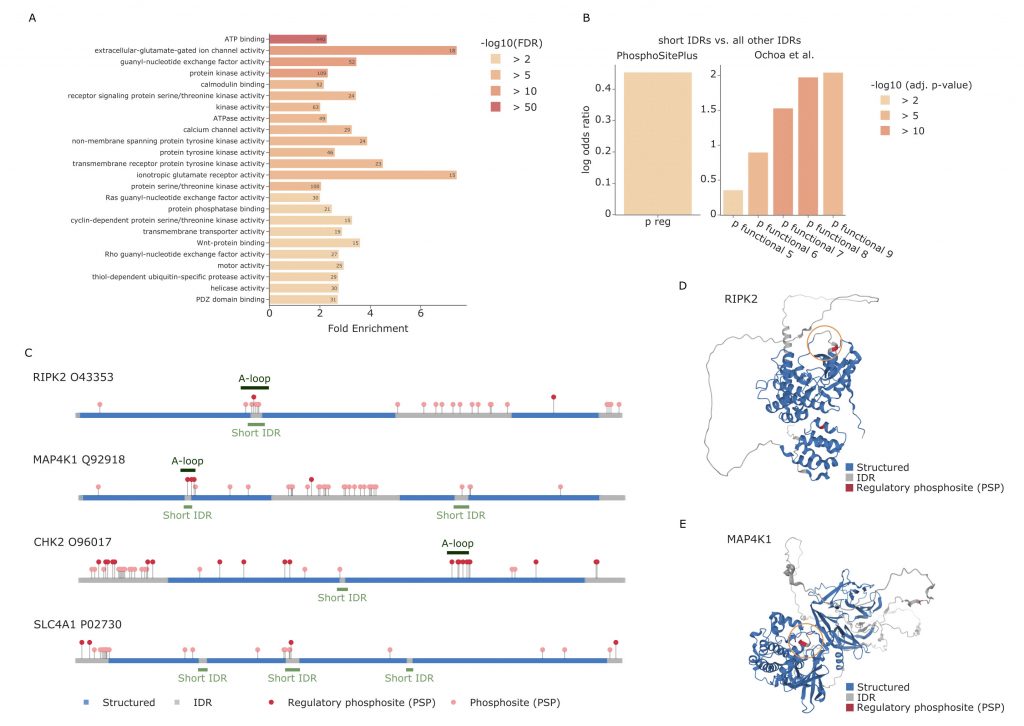
PTMs proximity in 3D
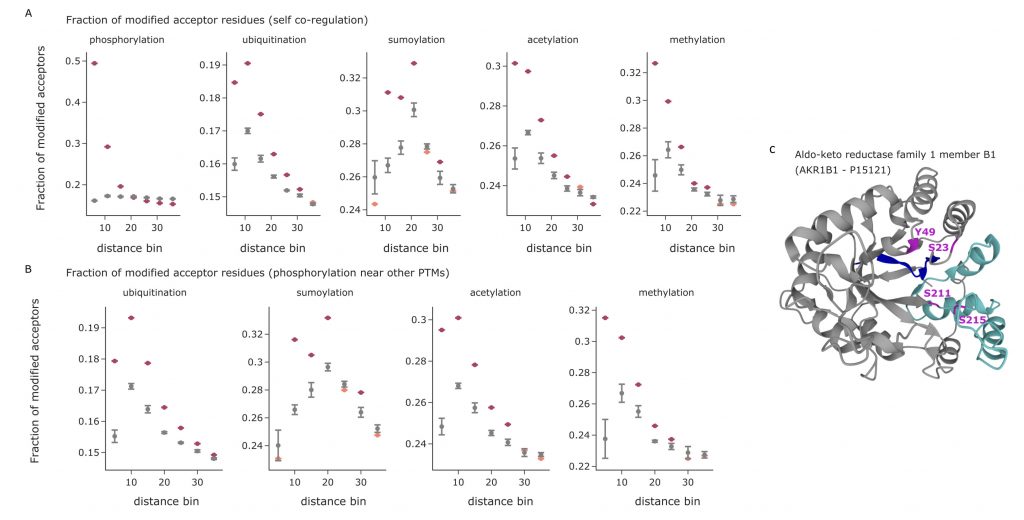
It has been proven that PTM-modified sites tend to induce other modifications in regions of the sequence near to the originally modified site and that multiple modifications in sequence proximity are key to regulatory functions and binding properties. Thanks to the structures provided by AlphaFold, the proximity between amino acids can now be defined not only in terms of sequence distance, but also in terms of 3D-proximity. The authors calculate the 3D distance between the various annotated modifications in structured regions and short IDRs, while discarding IDRs since they introduce greater structural uncertainty. For each modification type, the distances to the modifications of the same type were measured and compared to the distances that can be obtained from 5 identical structures with randomly permuted PTMs sites. The results show that PTMs indeed tend to cluster together, with preferential modifications of the acceptors situated near an already modified site. The same analysis was repeated to measure the proximity between different PTM types, observing similar preferential modifications. As an example, it is possible to observe that ubiquitinations, acetylations, and methylations seem to preferentially take place near already modified phosphosites.
Conclusion
The work presented here illustrates how combining mass spectrometry data with structural information helps to gain further insight into PTMs’ regulatory role. Studying PTMs considering their localization in proteins can unveil patterns that are not visible without structural information. This article is a great example of how the interpretation of mass-spectrometry data can be extended by merging the information with other types of data.
Bibliography:
Hochstrasser, M., 1996. UBIQUITIN-DEPENDENT PROTEIN DEGRADATION. Annual Review of Genetics 30, 405–439.. doi:10.1146/annurev.genet.30.1.405h
Millán-Zambrano, G., Burton, A., Bannister, A.J. et al. Histone post-translational modifications — cause and consequence of genome function. Nat Rev Genet (2022). https://doi.org/10.1038/s41576-022-00468-7
Bludau, I., Willems, S., Zeng, W.-F., Strauss, M. T., Hansen, F. M., Tanzer, M. C., Karayel, O., Schulman, B. A., & Mann, M. (2022). The structural context of PTMs at a proteome wide scale. Cold Spring Harbor Laboratory. https://doi.org/10.1101/2022.02.23.481596
Jumper, J., Evans, R., Pritzel, A. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). https://doi.org/10.1038/s41586-021-03819-2
Varadi, M. et al., AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models, Nucleic Acids Research, Volume 50, Issue D1, 7 January 2022, Pages D439–D444, https://doi.org/10.1093/nar/gkab1061

Categories
Latest posts

08/05/2024 - Journal Club
Cross-Border Collaboration: Enhancing Peptide Identification with MS2Rescore and MS Amanda

08/09/2023 - Journal Club
Exploring Cellular Complexity: Unveiling Single-Cell Proteomics

23/08/2023 - Journal Club
Modeling Lower-Order Statistics to Enable Decoy-Free FDR Estimation in Proteomics